Photo from wikipedia
Lysosomal storage disorders (LSDs) are inherited metabolic diseases caused by genetic defects in lysosomal enzymes or related factors. LSDs are associated with excessive accumulation of natural substrates in lysosomes leading… Click to show full abstract
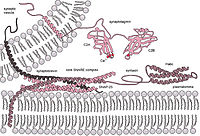
Lysosomal storage disorders (LSDs) are inherited metabolic diseases caused by genetic defects in lysosomal enzymes or related factors. LSDs are associated with excessive accumulation of natural substrates in lysosomes leading to central nervous system and peripheral tissue damage. Abnormal autophagy is also involved in pathogenesis, although the underlying mechanisms remain unclear. We demonstrated that impairment of lysosome‐autophagosome fusion is due to suppressed endocytosis in LSDs. The fusion was reduced in several LSD cells and the brains of LSD model mice, suggesting that the completion of autophagy is suppressed by the accumulation of substrates. In this brain, the expression of the soluble N‐ethylmaleimide sensitive factor attachment protein receptor (SNARE) proteins, VAMP8 and Syntaxin7, was decreased on the lysosomal surface but not intracellular. This aberrant autophagy preceded the development of pathological phenotypes in LSD‐model mice. Furthermore, the enzyme deficiency leading to the substrate accumulation could suppress endocytosis, and the inhibited endocytosis decreased SNARE proteins localized on lysosomes. These findings suggest that the shortage of SNARE proteins on lysosomes is one of the reasons for the impairment of lysosome‐autophagosome fusion in LSD cells. Defects in lysosomal enzyme activity suppress endocytosis and decrease the supply of intracellular SNARE proteins recruited to lysosomes. This shortage of lysosomal SNARE proteins impairs lysosome‐autophagosome fusion in lysosomal storage disorders.
Journal Title: Journal of Inherited Metabolic Disease
Year Published: 2022
Link to full text (if available)
Share on Social Media: Sign Up to like & get
recommendations!