Photo from wikipedia
Arginase 1 Deficiency (ARG1‐D) is a rare urea cycle disorder that results in persistent hyperargininemia and a distinct, progressive neurologic phenotype involving developmental delay, intellectual disability, and spasticity, predominantly affecting… Click to show full abstract
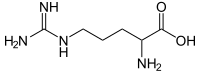
Arginase 1 Deficiency (ARG1‐D) is a rare urea cycle disorder that results in persistent hyperargininemia and a distinct, progressive neurologic phenotype involving developmental delay, intellectual disability, and spasticity, predominantly affecting the lower limbs and leading to mobility impairment. Unlike the typical presentation of other urea cycle disorders, individuals with ARG1‐D usually appear healthy at birth and hyperammonemia is comparatively less severe and less common. Clinical manifestations typically begin to develop in early childhood in association with high plasma arginine levels, with hyperargininemia (and not hyperammonemia) considered to be the primary driver of disease sequelae. Nearly five decades of clinical experience with ARG1‐D and empirical studies in genetically manipulated models have generated a large body of evidence that, when considered in aggregate, implicates arginine directly in disease pathophysiology. Severe dietary protein restriction to minimize arginine intake and diversion of ammonia from the urea cycle are the mainstay of care. Although this approach does reduce plasma arginine and improve patients' cognitive and motor/mobility manifestations, it is inadequate to achieve and maintain sufficiently low arginine levels and prevent progression in the long term. This review presents a comprehensive discussion of the clinical and scientific literature, the effects and limitations of the current standard of care, and the authors' perspectives regarding the past, current, and future management of ARG1‐D.
Journal Title: Journal of Inherited Metabolic Disease
Year Published: 2022
Link to full text (if available)
Share on Social Media: Sign Up to like & get
recommendations!