Photo from wikipedia
The equilibrium between histone acetylation and deacetylation plays an important role in cancer initiation and progression. The histone deacetylases (HDACs) are a class of key regulators of gene expression that… Click to show full abstract
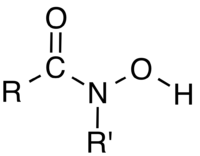
The equilibrium between histone acetylation and deacetylation plays an important role in cancer initiation and progression. The histone deacetylases (HDACs) are a class of key regulators of gene expression that enzymatically remove an acetyl moiety from acetylated lysine ε-amino groups on histone tails. Therefore, HDAC inhibitors have recently emerged as a promising strategy for cancer therapy and several pan-HDAC inhibitors have globally been approved for clinical use. In the present study, we designed and synthesized a series of substituted indole-based hydroxamic acid derivatives that exhibited potent anti-proliferative activities in various tumor cell lines. Among the compounds tested, compound 4o, was found to be among the most potent in the inhibition of HDAC1 (half maximal inhibitory concentration, IC50 = 1.16 nM) and HDAC6 (IC50 = 2.30 nM). It also exhibited excellent in vitro anti-tumor proliferation activity. Additionally, compound 4o effectively increased the acetylation of histone H3 in a dose-dependent manner and inhibited cell proliferation by inducing cell cycle arrest and apoptosis. Moreover, compound 4o remarkably blocked colony formation in HCT116 cancer cells. Based on its favorable in vitro profile, compound 4o was further evaluated in an HCT116 xenograft mouse model, in which it demonstrated better in vivo efficacy than the clinically used HDAC inhibitor, suberanilohydroxamic acid. Interestingly, compound 4k was found to have a preference for the inhibition of HDAC6, with IC50 values of 115.20 and 5.29 nM against HDAC1 and HDAC6, respectively.
Journal Title: European journal of medicinal chemistry
Year Published: 2021
Link to full text (if available)
Share on Social Media: Sign Up to like & get
recommendations!