Photo from wikipedia
ABSTRACT Purpose: To expand the genotype/phenotype correlations in patients with autosomal dominant retinitis pigmentosa (adRP) harboring PRPF8 variants. Materials and Methods: Two patients, a father and his daughter, harboring a… Click to show full abstract
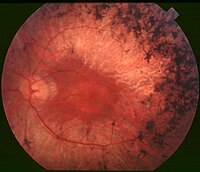
ABSTRACT Purpose: To expand the genotype/phenotype correlations in patients with autosomal dominant retinitis pigmentosa (adRP) harboring PRPF8 variants. Materials and Methods: Two patients, a father and his daughter, harboring a novel p.PRPF8-Glu2331* variant, underwent ophthalmic examination at 3-year-interval, including fundus photography, fundus autofluorescence, optical coherence tomography, and ISCEV standard full field ERGs. All reported disease-causing PRPF8 variants were collected and localized in the PRPF8 and PRPF8/SNRNP200 protein structures. Results: The p.PRPF8-Glu2331* variant results in a truncated PRPF8 protein lacking the last five C-terminal amino acids and caused in the two patients a severe clinical phenotype, with the macula being affected from the second decade on. All but two adRP-linked variants are located in the last exon 43 encoding the C-terminal tail of the C-terminal PRPF8 Jab1 domain. The p.PRPF8-Ser2118Phe and -Asn2280Lys variants encoded by exons 39 and 42, respectively, are located at the basis of the C-terminal tail. Conclusions: Frame-shift mutations and nonconservative amino acid changes in PRPF8 typically cause severe clinical phenotypes. The conservative missense variant p.PRPF8-Arg2310Lys that is not altering the global charge of the C-terminal tail, and variants located at the basis of the C-terminal tail show milder clinical phenotypes, in accordance with functional data on PRPF8/SNRNP200 interactions in yeast.
Journal Title: Ophthalmic Genetics
Year Published: 2018
Link to full text (if available)
Share on Social Media: Sign Up to like & get
recommendations!