Photo from wikipedia
Background Illumina technology currently dominates bacterial genomics due to its high read accuracy and low sequencing cost. However, the incompleteness of draft genomes generated by Illumina reads limits their application… Click to show full abstract
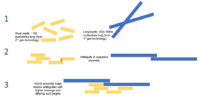
Background Illumina technology currently dominates bacterial genomics due to its high read accuracy and low sequencing cost. However, the incompleteness of draft genomes generated by Illumina reads limits their application in comprehensive genomics analyses. Alternatively, hybrid assembly using both Illumina short reads and long reads generated by single molecule sequencing technologies can enable assembly of complete bacterial genomes, yet the high per-genome cost of long-read sequencing limits the widespread use of this approach in bacterial genomics. Here we developed a protocol for hybrid assembly of complete bacterial genomes using miniaturized multiplexed Illumina sequencing and non-barcoded PacBio sequencing of a synthetic genomic pool (SGP), thus significantly decreasing the overall per-genome cost of sequencing. Results We evaluated the performance of SGP hybrid assembly on the genomes of 20 bacterial isolates with different genome sizes, a wide range of GC contents, and varying levels of phylogenetic relatedness. By improving the contiguity of Illumina assemblies, SGP hybrid assembly generated 17 complete and 3 nearly complete bacterial genomes. Increased contiguity of SGP hybrid assemblies resulted in considerable improvement in gene prediction and annotation. In addition, SGP hybrid assembly was able to resolve repeat elements and identify intragenomic heterogeneities, e.g. different copies of 16S rRNA genes, that would otherwise go undetected by short-read-only assembly. Comprehensive comparison of SGP hybrid assemblies with those generated using multiplexed PacBio long reads (long-read-only assembly) also revealed the relative advantage of SGP hybrid assembly in terms of assembly quality. In particular, we observed that SGP hybrid assemblies were completely devoid of both small (i.e. single base substitutions) and large assembly errors. Finally, we show the ability of SGP hybrid assembly to differentiate genomes of closely related bacterial isolates, suggesting its potential application in comparative genomics and pangenome analysis. Conclusion Our results indicate the superiority of SGP hybrid assembly over both short-read and long-read assemblies with respect to completeness, contiguity, accuracy, and recovery of small replicons. By lowering the per-genome cost of sequencing, our parallel sequencing and hybrid assembly pipeline could serve as a cost effective and high throughput approach for completing high-quality bacterial genomes.
Journal Title: BMC Genomics
Year Published: 2020
Link to full text (if available)
Share on Social Media: Sign Up to like & get
recommendations!