Photo from wikipedia
The presence of N-nitrosodimethylamine (NDMA) and N-nitrosodiethylamine (NDEA) impurities in angiotensin II receptor blocker (ARB) drugs containing tetrazole ring has triggered worldwide product recalls. The purpose of this article is… Click to show full abstract
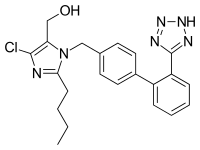
The presence of N-nitrosodimethylamine (NDMA) and N-nitrosodiethylamine (NDEA) impurities in angiotensin II receptor blocker (ARB) drugs containing tetrazole ring has triggered worldwide product recalls. The purpose of this article is to identify the potential gap area in current pharmaceutical industry practice that might have led to the NMDA and NDEA impurities escaping the drug manufacturer’s and FDA’s attention. The impact of process change was not adequately assessed by the manufacturer of contaminated APIs (active pharmaceutical ingredients), and potential for generation of mutagenic or other toxic impurities was not considered. The safety and risk associated with a chemical synthetic process was also not evaluated. This is primarily due to current industry practice which focuses on controlling the impurities above reporting threshold. ICH Q3A and FDA guidance on genotoxic and carcinogenic impurities in drug substances and products need to be integrated so that the ICH Q3A decision tree (attachment 3) begins by checking whether the synthetic process has been evaluated for the potential to generate toxic impurities. The compliance with ICH Q3A limits should be carried out only after the process has been determined to be safe without the risk of generating mutagenic and carcinogenic impurities.
Journal Title: AAPS PharmSciTech
Year Published: 2019
Link to full text (if available)
Share on Social Media: Sign Up to like & get
recommendations!