Photo from wikipedia
AIM To discover the molecular pathogenic basis of the blepharophimosis, ptosis, and epicanthus inversus syndrome (BPES), and to predict the clinical subtype according to in vitro experiments, which is significant… Click to show full abstract
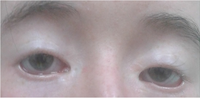
AIM To discover the molecular pathogenic basis of the blepharophimosis, ptosis, and epicanthus inversus syndrome (BPES), and to predict the clinical subtype according to in vitro experiments, which is significant to the prognosis. METHODS A 3-year-old sporadic female patient with typical clinical manifestations of BPES was enrolled. The coding region of forkhead box L2 (FOXL2) gene was sequenced, and the functional assays were performed in vitro by Western blotting, subcellular localization experiment, luciferase reporter assay, and quantitative real-time polymerase chain reaction. RESULTS A novel FOXL2 point pathogenic variant (c.274G>T) was detected, resulting in a truncated protein (p.E92*). Functional studies demonstrated that the FOXL2 pathogenic variant induced the subcellular mislocalization and the abnormal transcriptional activity on promoters of the steroidogenic acute regulatory protein (StAR or STARD1) gene and the odd-skipped related 2 transcription factor (OSR2) gene. CONCLUSION A novel pathogenic variant is identified to expand the spectrum of the known FOXL2 mutations. The in vitro experiments provide reference data and more insights to the molecular pathogenesis of BPES. The predicted high risk of ovarian insufficiency makes it significant for the patient enrolled to have further follow-up and therapy concerning female endocrinology.
Journal Title: International journal of ophthalmology
Year Published: 2023
Link to full text (if available)
Share on Social Media: Sign Up to like & get
recommendations!