Photo from wikipedia
TMBIM5 deficiency reduces mitochondrial K+/H+ exchange. Mutation of the channel pore in mice destabilizes the protein and results in increased embryonic lethality and a skeletal myopathy. Ion fluxes across the… Click to show full abstract
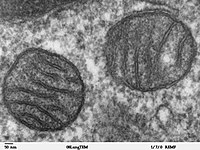
TMBIM5 deficiency reduces mitochondrial K+/H+ exchange. Mutation of the channel pore in mice destabilizes the protein and results in increased embryonic lethality and a skeletal myopathy. Ion fluxes across the inner mitochondrial membrane control mitochondrial volume, energy production, and apoptosis. TMBIM5, a highly conserved protein with homology to putative pH-dependent ion channels, is involved in the maintenance of mitochondrial cristae architecture, ATP production, and apoptosis. Here, we demonstrate that overexpressed TMBIM5 can mediate mitochondrial calcium uptake. Under steady-state conditions, loss of TMBIM5 results in increased potassium and reduced proton levels in the mitochondrial matrix caused by attenuated exchange of these ions. To identify the in vivo consequences of TMBIM5 dysfunction, we generated mice carrying a mutation in the channel pore. These mutant mice display increased embryonic or perinatal lethality and a skeletal myopathy which strongly correlates with tissue-specific disruption of cristae architecture, early opening of the mitochondrial permeability transition pore, reduced calcium uptake capability, and mitochondrial swelling. Our results demonstrate that TMBIM5 is an essential and important part of the mitochondrial ion transport system machinery with particular importance for embryonic development and muscle function.
Journal Title: Life Science Alliance
Year Published: 2022
Link to full text (if available)
Share on Social Media: Sign Up to like & get
recommendations!