Photo from wikipedia
Whole exome sequencing (WES) of matched tumor-normal pairs in rare tumors has the potential to identify genome-wide mutations and copy number alterations (CNAs). We evaluated 27 rare cancer patients with… Click to show full abstract
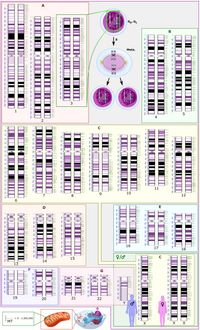
Whole exome sequencing (WES) of matched tumor-normal pairs in rare tumors has the potential to identify genome-wide mutations and copy number alterations (CNAs). We evaluated 27 rare cancer patients with tumor-normal matching by WES and tumor-only next generation sequencing (NGS) as a comparator. Our goal was to: 1) identify known and novel variants and CNAs in rare cancers with comparison to common cancers; 2) examine differences between germline and somatic variants and how that functionally impacts rare tumors; 3) detect and characterize alleles in biologically relevant genes-pathways that may be of clinical importance but not represented in classical cancer genes. We identified 3343 germline single nucleotide variants (SNVs) and small indel variants—1670 in oncogenes and 1673 in tumor suppressor genes—generating an average of 124 germline variants/case. The number of somatic SNVs and small indels detected in all cases was 523:306 in oncogenes and 217 in tumor suppressor genes. Of the germline variants, six were identified to be pathogenic or likely pathogenic. In the 27 analyzed rare cancer cases, CNAs are variable depending on tumor type, germline pathogenic variants are more common. Cell fate pathway mutations (e.g., Hippo, Notch, Wnt) dominate pathogenesis and double hit (mutation + CNV) represent ~18% cases.
Journal Title: Cancers
Year Published: 2020
Link to full text (if available)
Share on Social Media: Sign Up to like & get
recommendations!