Photo from wikipedia
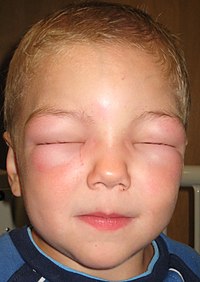
This review aims to summarize the main pathophysiological events involved in the development of hereditary angioedema (OMIM#106100). Hereditary angioedema is a rare genetic disease inherited in an autosomal dominant manner… Click to show full abstract
This review aims to summarize the main pathophysiological events involved in the development of hereditary angioedema (OMIM#106100). Hereditary angioedema is a rare genetic disease inherited in an autosomal dominant manner and caused by a loss of control over the plasma contact system or kallikrein-kinin system, which results in unrestrained bradykinin generation or signaling. In patients with hereditary angioedema, BK binding to endothelial cells leads to recurrent episodes of swelling at subcutaneous or submucosal tissues that can be life threatening when affecting the upper respiratory tract. The disease can either present with hypocomplementemia owing to the presence of pathogenic variants in the gene encoding complement C1 inhibitor (hereditary angioedema with C1-inhibitor deficiency) or present with normocomplementemia and associate with elevated estrogen levels owing to gain-of-function variants in the genes encoding coagulation proteins involved in the kallikrein-kinin system (namely, coagulation FXII [FXII-associated hereditary angioedema], plasminogen [PLG-associated hereditary angioedema], and high-molecular-weight kininogen [KNG1-associated hereditary angioedema]). Moreover, in recent years, novel pathogenic variants have been described in the genes encoding angiopoietin 1 (ANGPT1-associated hereditary angioedema) and myoferlin (MYOF-associated hereditary angioedema), which further expand the pathophysiological picture of hereditary angioedema.
Journal Title: Balkan Medical Journal
Year Published: 2020
Link to full text (if available)
Share on Social Media: Sign Up to like & get
recommendations!